Hora da genética!
Confira nossas publicações sobre doenças genéticas.

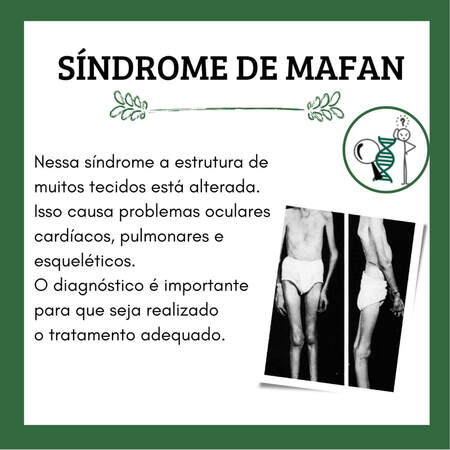 A Síndrome de Marfan (SMF) é uma doença autossômica hereditária causada por mutações dos genes envolvidos na codificação da proteína fibrila. Essa proteína é um importante componente da matriz de tecidos elásticos e não elásticos, o que leva à alterações de tecido conectivo.
O primeiro relato de uma mutação no gene FBN1 ocorreu em 1991, desde então, mais de 1.800 mutações diferentes envolvendo essa proteína foram registradas para MFS e distúrbios associados. O FBN1 é um gene grande (65 éxons) localizado no cromossomo 15q21. Segundo estudos, a incidência relatada na população varia de 1 em 3.000 a 5.000 indivíduos e a gravidade clínica associada à SMF é bastante variável, desde características isoladas à apresentação neonatal de doença grave e rapidamente progressiva, que podem envolver múltiplos sistemas. Algumas manifestações que podem ser encontradas na SMF, são: doença aórtica, doença cardíaca, alterações esqueléticas, alterações oculares, doença pulmonar, entre outras. Em relação a doença aórtica, nesta síndrome pode ocorrer dilatação aneurismática, regurgitação aórtica e dissecção, sendo esta causa o principal fator de morbidade e mortalidade. A dilatação aórtica é encontrada em cerca de 50% das crianças e 80% dos adultos com SMF. Por isso, quando a síndrome não é diagnosticada e não é instituído tratamento, o desfecho geralmente é de dissecção da aorta. Dos achados esqueléticos, destaca-se o crescimento linear excessivo dos ossos longos e a frouxidão articular. Em geral, os indivíduos com SMF são mais altos do que o esperado para seu histórico genético. Outras características bastante encontradas são: aracnodactilia (dedos longos, delgados e curvos); pectus carinatum e pectus excavatum (alterações no formato do osso esterno); e escoliose e cifose (curvaturas anormais da coluna vertebral). As alterações oculares são comuns nos pacientes com SMF, por isso, é recomendada avaliação oftalmológica anual. Estima-se que a ectopia lentis - uma subluxação do cristalino - ocorra em 50 a 80% dos indivíduos com a síndrome. As alterações pulmonares estão relacionadas com presença de características enfisematosas e predisposição a pneumotórax espontâneo. Sobre o diagnóstico desta síndrome, em geral, usa-se os Critérios Ghent revisados de 2010. Estes critérios são baseados na presença ou ausência de história familiar (parentes de primeiro ou segundo grau), exame físico (alterações esqueléticas, oculares, características faciais entre outras), imagem da aorta e testes genéticos em alguns casos. O diagnóstico diferencial inclui diversas condições que possuem características fenotípicas semelhantes ao fenótipo de Marfan, como outras doenças associadas a mutações dos genes FBN1/2 ou TGFBR1/2. Em muitos casos não é possível a exclusão da SMF em indivíduos menores de 20 anos. Assim, na presença de achados sistêmicos sugestivos, mas sem envolvimento cardiovascular, é necessário o acompanhamento desses indivíduos com ecocardiogramas anuais até pelo menos 20 anos de idade ou pararem de crescer. Para os adultos com medições repetidamente normais da raiz da aorta, recomenda-se acompanhamento em intervalos de dois a três anos. É importante destacar que as diretrizes de 2010 do American College of Cardiology/American Heart Association/American Association for Thoracic Surgery, recomendam a triagem de parentes dos pacientes com SMF. Desta forma, os parentes de primeiro grau de pacientes que tenham mutação genética associada a aneurismas e/ou dissecção aórtica devem passar por aconselhamento e testes genéticos. Por fim, com o avanço dos estudos e o diagnóstico oportuno, o prognóstico dos pacientes com SMF tem melhorado. Nesse sentido, é imprescindível o uso da terapia medicamentosa (betabloqueadores e bloqueadores dos receptores de angiotensina), monitoramento rotineiro e não invasivo do tamanho da aorta, reparo cirúrgico eletivo da aorta e restrição de exercícios físicos vigorosos. Além disso, devido às alterações fisiológicas que ocorrem durante a gravidez, as pacientes com SMF requerem monitoramento mais intensivo pelo risco aumentado de dilatação e dissecção aórtica. Angélica N. P. Schindler Discente de Medicina - UNILA REFERÊNCIAS WRIGHT, M. J.; CONNOLLT, H. M. Genetics, clinical features, and diagnosis of Marfan syndrome and related disorders. UpToDate: out, 2022. WRIGHT, M. J.; CONNOLLT, H. M. Management of Marfan syndrome and related disorders. UpToDate: out, 2022. ARAÚJO, M. R. et al. Síndrome de Marfan: novos critérios diagnósticos, mesma abordagem anestésica? Relato de caso e revisão. Brazilian Journal of Anesthesiology, v. 66, (4,) jul-ago 2016, p. 408-413. Disponível em: https://doi.org/10.1016/j.bjan.2016.04.002.
0 Comments
Leave a Reply. |
AutorEscreva algo sobre si mesmo. Não precisa ser extravagante, apenas uma visão geral. Histórico
Maio 2022
Categorias |
 Feed RSS
Feed RSS
