Dúvidas sobre
doenças genéticas?
Pergunte que eu respondo!

 A variabilidade genética refere-se as diferenças entre indivíduos ou diferenças entre populações. As mutações são a causa de base da variabilidade genética, mas mecanismos como a reprodução sexual e à deriva genética também contribuem para isso. A variabilidade no genoma humano pode se apresentar de diferentes maneiras uma delas são os polimorfismos e em outras ocasiões, as variações ocorrem em uma escala maior, por exemplo, quando um segmento de DNA de centenas ou milhares de pares de bases é diferente entre as pessoas.
O DNA de todas as espécies conhecidas de organismos tem a mesma estrutura química; entretanto, cada organismo é completamente diferente do outro; a diferença se deve à ordem das bases nitrogenadas na molécula de DNA. Organismos da mesma espécie compartilham sequências em sua molécula de DNA, mas, mesmo dentro da mesma espécie existem variações entre os indivíduos. Organismos da mesma espécie compartilham regiões de sua sequência em até pouco mais de 99%, o que lhes confere características muito semelhantes. Além disso, parentes próximos terão sequências com maior similaridade entre si, mas nunca serão as mesmas, o que define a variabilidade genética intra e interespécies. No caso do DNA humano, as sequências que contêm os genes não são muito variáveis dentro da espécie; No entanto, o restante da sequência é muito sujeito à variabilidade e, como existem milhões de pares de bases por molécula de DNA e uma alta porcentagem deles codificam alguma proteína, cada pessoa tem uma sequência única de DNA o que permite que isso aconteça identificado apenas pela ordem de seus pares de bases. O que são os polimorfismos? Polimorfismo significa literalmente "muitas formas", se a variação é encontrada em uma frequência superior a 1% da população, se denomina polimorfismo. Os polimorfismos são variantes do genoma que aparecem por meio de mutações em alguns indivíduos e são transmitidos aos descendentes pelo que adquirem certa frequência na população após múltiplas gerações. Estima-se que exista uma variante em cada 1.000 pares de bases dos 3.000 milhões que constituem o genoma humano. Os polimorfismos são a base da evolução e aqueles que se consolidam podem ser silenciosos ou trazer vantagens para os indivíduos, embora também possam contribuir para causar doenças. Muitas doenças determinadas geneticamente pelas chamadas mutações ou variantes de "alta penetrância" são conhecidas, uma vez que os portadores da variante tendem a manifestar a doença com alta probabilidade. Assim, o polimorfismo genético, cromossômico ou de sequência de DNA são responsáveis pela grande variabilidade existente entre indivíduos de uma mesma espécie. Eles podem atuar como marcadores genéticos, já que são transmitidos e associados a outros genes localizados na região cromossômica próxima a eles. Desta forma, se um gene próximo a um marcador causa uma doença, todos os indivíduos afetados na família recebem tanto o marcador como o gene causador da doença. Essas variantes tendem a ser de baixa frequência na população em geral, por exemplo, mutações herdadas no gene supressor de tumor APC determinam o aparecimento de Polipose Adenomatosa Familiar que muitas vezes evolui em carcinoma no cólon, mas essa entidade não explica mais do que 1% de todos os tumores de cólon. Além disso, diferentes fenótipos são decorrentes de alguns polimorfismos, como, por exemplo, o sistema ABO. Os polimorfismos podem ter significados funcionais diferentes, dependendo se eles afetam uma região codificadora do genoma, uma região reguladora ou uma região não codificadora. Os polimorfismos nas regiões codificantes são chamados de "polimorfismos genéticos". Esta classe pode ou não ter efeito sobre o fenótipo. Polimorfismos gênicos, sem efeito fenotípico, são os mais comuns e são responsáveis pela diversidade genética normal entre os indivíduos (por exemplo, polimorfismos existentes em proteínas plasmáticas, como imunoglobulinas). Mas quando um polimorfismo gênico (ou seja, um gene afetado na região do DNA codificador) resulta em uma alteração fenotípica, na maioria das vezes é prejudicial, pois pode modificar as características bioquímicas, fisiológicas e até morfológicas da célula, podendo originar processos patológicos. Apenas em casos excepcionais, essa variação ou mutação pode ser benéfica, dando origem a uma vantagem adaptativa ao indivíduo, sendo este o motor da evolução da espécie. Polimorfismos com alteração do fenótipo, mas que não influenciam a susceptibilidade a doenças, determinam as características diferenciais entre indivíduos de uma mesma espécie, como altura, cabelo e cor dos olhos, etc. Polimorfismos para fins diagnósticos A análise dos polimorfismos pode ser utilizada para detectar a predisposição a uma determinada doença, e até mesmo detectá-la antes de seu desenvolvimento. Isso é de grande interesse no diagnóstico pré-natal de doenças congênitas. A identificação precoce desse tipo de doença pode permitir o início de um tratamento preventivo o que é mais eficaz para atenuar as manifestações clínicas da doença em questão. Por outro lado, é possível estudar á herança e a relação das doenças genéticas com as famílias, e está sendo utilizada em doenças como fibrose cística e coreia de Huntington. A impressão digital genética pode ser utilizada em estudos para estabelecer relações de parentesco, em testes de paternidade, na identificação de recém-nascidos e em casos de criminologia, entre outros. O princípio básico deste método é conseguir examinar um número suficiente de polimorfismos, o que gera uma probabilidade menor de que um outro indivíduo tenha os mesmos alelos na sequência de DNA, logo comparam as amostras para encontrar por exemplo o responsável de um crime. Por: Catherine Molina, Acadêmica do curso de Medicina da UNILA. Referências: Herrera, Edwin. La genética de poblaciones y el origen de la diversidad humana. Revista médica de Honduras. Vol 81 (1), 2013. Disponível em: http://www.bvs.hn/RMH/pdf/2013/pdf/Vol81-1-2013-10.pdf Iniesta, Raquel; Guinó, Elisabet; Moreno, Víctor. Análisis estadístico de polimorfismos genéticos en estudios epidemiológico. Revista Gaceta Sanitaria. Vol.19 (4), Barcelona, España, 2005. Disponível em: https://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S0213-91112005000400011 Thompson, Margaret; Thompos, James. Genética Médica. Editorial Elsevier. 6° Edição, 2002. Torrades, Sandra. Diversidad del genoma humano: polimorfismos. Elsevier. Vol 21 (5), mayo 2002. Págs. 122-125. Disponível em: https://www.elsevier.es/es-revista-offarm-4-articulo-diversidad-del-genoma-humano-polimorfismos-13031745
0 Comments
As mitocôndrias são organelas celulares que possuem DNA próprio. Como via de regra, para os seres humanos, considera-se que a herança mitocondrial seja materna, ou seja, o DNA presente nas mitocôndrias (mtDNA) é herdado da mãe, visto que as mitocôndrias do espermatozoide são eliminadas no processo de fertilização e somente aquelas presentes no oócito são conservadas no zigoto (YAN, C. et al. 2019). No entanto, contrariando o dogma central da herança mitocondrial, os estudos de Luo et al. (2018), apontam para casos excepcionais, em que esse padrão de herança pode não ocorrer, necessariamente: a heteroplasmia. A heteroplasmia consiste na herança biparental do mtDNA, ou seja, o indivíduo apresenta tanto o genoma mitocondrial materno quanto o paterno, fazendo com que o padrão de herança seja afetado, a depender da análise do tecido em questão e da porcentagem expressa de mtDNA em cada indivíduo, interferindo categoricamente no aconselhamento genético, sendo que estudos pontuais, individuais e altamente refinados serão necessários para a análise de cada caso. Dessa maneira, a heteroplasmia foi identificada pelos pesquisadores ao estudar indivíduos suspeitos de sofrerem distúrbios mitocondriais. Assim, ao realizar o sequenciamento do material genético mitocondrial, puderam observar a presença do mtDNA de ambos os pais, expressa nos filhos e, inclusive, ao longo de toda a família, caracterizando essa herança como autossômica dominante. De acordo com os resultados, o heredograma a seguir, retirado do estudo em questão, apresenta o padrão de herança heteroplásmica de uma das famílias estudadas: 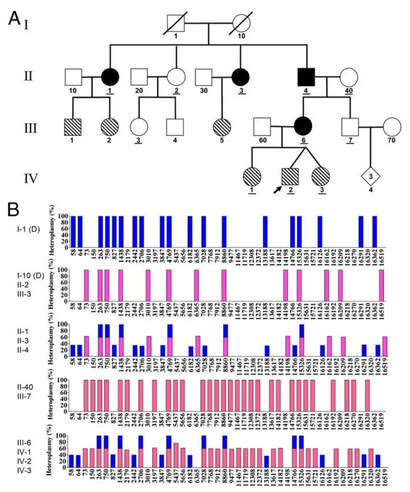 Luo et al. (2018) – modificado. No esquema A, os símbolos preenchidos em preto são os indivíduos que receberam a herança biparental mitocondrial. Já os preenchidos na diagonal indicam os indivíduos com alto nível de heteroplasmia, mas com herança materna normal. Os indivíduos sublinhados são os que foram sequenciados para o mtDNA e as entradas com “D” são genomas deduzidos. No esquema B, as barras azuis representam o genoma mitocondrial de origem paterna, já as barras em rosa e em vermelho, de origem materna.
Dessa forma, pode-se observar que o probando (IV-2) apresenta o mesmo mtDNA da mãe (III-6), a qual possui a heteroplasmia, visto que recebeu o mtDNA tanto de seu pai (II-4), quanto de sua mãe (II-40). Além disso, o heredograma também aponta para que esta seja uma herança autossômica dominante, na qual, uma vez que instalada, não tende a pular gerações. A partir do exposto, pode-se perceber que a herança mitocondrial se apresenta, na verdade, de uma forma muito mais complexa do que o clássico padrão apresentado até então. Cabe aos pesquisadores, portanto, compreender a gama de informações que vem se apresentando, para que no futuro, novas técnicas de reconhecimento, terapia e tratamento possam ser desenvolvidas à luz dos novos conhecimentos adquiridos, podendo vir a corroborar, inclusive, nos processos de aconselhamento genético que envolvam as doenças de origem mitocondrial. Por: Camila Begui, Bióloga e Acadêmica do curso de Medicina da UNILA. REFERÊNCIAS: LUO, S. et al. Biparental Inheritance of Mitochondrial DNA in Humans. Proceedings of the National Academy of Sciences, v. 115, n. 51, p. 13039-13044, dez. 2018. Disponível em: https://www.pnas.org/content/115/51/13039. Acesso em: 24 de agosto de 2021. YAN, C. et al. Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells, v. 8, n. 4, p. 379-394, abr. 2019. Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6523345/. Acesso em: 24 de agosto de 2021. Existe uma mutação genética descoberta no ano 1994 chamada: Fator V de Leiden. Essa mutação predispõe os portadores ao tromboembolismo hereditário, ao qual gera uma desordem multifatorial que envolve a interação de fatores de risco genéticos e/ou adquiridos que afetam as proteínas do sistema anticoagulante. Dentre esses fatores, as mutações nos genes do fator V e da protrombina são as duas causas que prevalecem para o desenvolvimento da trombose hereditária.
Para entendermos um pouco melhor, é importante explicar o que é o fator V: é uma proteína plasmática formada por 2.196 aminoácidos, precursor do fator Va (fator V ativado) essencial para a síntese da trombina, responsável pela ativação da coagulação. O processo de desativação baseia-se na ligação da trombina e da proteína C à trombomodulina (presente no endotélio) convertendo a proteína C à proteína C ativada (PCA). Esta ativação ocorre na presença da proteína S (PS). O complexo PCA/ PS causa a inativação através da clivagem enzimática dos cofatores Va e VIIIa que inibem a atividade do sistema coagulante. A resistência à proteína C ativada é um dos principais fatores de risco para o tromboembolismo venoso e é a mutação do fator V de Leiden que representa uma das causas principais de resistência à proteína C. Cerca de 90% dos casos de resistência a esta proteína se devem à mutação de ponto no gene do fator V, da coagulação. Esta mutação ocorre no exon 10 do gene do fator V ocasionando uma substituição da base G/A (Guanina/Adenina) no nucleotídio 1691, resultando na troca da Arg (Arginina) pela Gln (Glutamina) na posição 506 da proteína, um dos principais sítios de clivagem para ativação da proteína C. A presença da mutação aumenta o risco de doença trombótica de três a dez vezes para portadores heterozigotos e de oitenta vezes para portadores homozigotos. Estudos mostram que a prevalência desta mutação dependerá da etnia da população estudada. A taxa da mutação dentro da população européia é de 5%, nos Estados Unidos é de 6% e praticamente ausente entre os africanos e asiáticos. Em afro-americanos é encontrada em cerca de 1%. No Brasil, a mutação está presente em cerca de 2% da população. Em pacientes portadores de doenças trombóticas, o fator V de Leiden foi observado em 20% dos casos. As técnicas de biologia molecular tornam possível a detecção de mutações, que podem ser realizadas, atualmente, como rotina laboratorial. A aplicação de teste de rastreamento destas mutações em pessoas com antecedentes clínicos e/ou familiar são de grande importância, pois permitem uma abordagem clínica antitrombótica, de modo a diminuir os riscos destas doenças. Os pacientes com doenças trombóticas (trombose venosa profunda, embolia pulmonar, trombose arterial e trombose cerebral vascular), podem ser analisados juntamente com sua família para detecção da distribuição dos alelos mutados a fim de correlacionar a presença da mutação e a sintomatologia. Finalmente, a detecção do fator V de Leiden em pacientes portadores de eventos trombóticos é recomendada para o esclarecimento das causas e para efetuar o rastreamento em membros de sua família, ainda sem o aparecimento de eventos trombóticos, de forma a avaliar os riscos associados e assim determinar um acompanhamento médico preventivo. Por: Nicolas Guzman, Acadêmico do curso de Medicina da UNILA. REFERÊNCIAS: Carvalho, Eunice B. et al. Rastreamento familiar do fator V de Leiden: a importância da detecção de portadores heterozigotos. Revista Brasileira de Hematologia e Hemoterapia [online]. 2005, v. 27, n. 2, pp. 83-86. Disponível em: <https://doi.org/10.1590/S1516-84842005000200005>. Epub 11 Jan 2006. ISSN 1806-0870. https://doi.org/10.1590/S1516-84842005000200005. Godoy, José M. P.Fator V de Leiden. Revista Brasileira de Hematologia e Hemoterapia [online]. 2005, v. 27, n. 2, pp. 79. Disponível em: <https://doi.org/10.1590/S1516-84842005000200001>. Epub 11 Jan 2006. ISSN 1806-0870. https://doi.org/10.1590/S1516-84842005000200001  O Transtorno do Espectro Autista (TEA) é um transtorno que engloba o comportamento e cognição, geralmente com início antes dos 3 anos de idade, afetando os domínios fundamentais da linguagem e desenvolvimento social com comportamentos repetitivos e restritivos (COUTINHO; BOSSO, 2015). O TEA é classificado como multicausal, no entanto, existem duas vertentes principais do autismo: o sindrômico e não sindrômico.
Estudos familiares e em gêmeos evidenciam a etiologia genética do autismo, mostrando a existência de um risco aumentado de recorrência do autismo de 3 a 8% em famílias com uma criança autista e concordância para o diagnóstico de autismo em gêmeos monozigóticos de pelo menos 71% (SOLÍS- AÑES, 2007; DELGADO-LUENGO, 2007; HERNÁNDEZ, 2007; GARDIA, 2004; TUCHMAN, 2004; ROTTA, 2004 apud COUTINHO; BOSSO, 2015). Embora o autismo pareça ser altamente hereditário, sua etiologia genética tem se mostrado bastante complexa, provavelmente envolvendo muitos genes em diferentes cromossomos atuando com efeito moderado (SOLÍS-AÑEZ, 2007; DELGADO- LUENGO, 2007; HERNÁNDEZ, 2007; GESCHWIND, 2008 apud COUTINHO; BOSSO, 2015). Existem autores que chegaram a afirmar que anomalias de quase todos os cromossomos já foram associadas ao autismo. Não implicando em um modelo próprio de transmissão genética ou um gene principal facilmente identificável como causa de desordens (GESCHWIND,2008 apud COUTINHO; BOSSO, 2015). Estudos de genética humana indicam que os genes da família SHANK podem estar envolvidos no autismo idiopático ou não sindrômico. Mutações nesses genes causam uma disfunção sináptica, a qual leva ao comportamento autista. A primeira triagem do genoma para regiões cromossômicas envolvidas no autismo associou aproximadamente 354 marcadores genéticos, localizados em oito regiões dos seguintes cromossomos: 2, 4, 7, 10, 13, 16, 19 e 22 sendo as regiões 7q, 16p, 2q, 17q mais relevantes (COUTINHO; BOSSO, 2015). As regiões do genoma humano 6q27, 20p13, e 5p15 foram descritas como regiões associadas com o autismo (WEISS et al. 2009 apud FREITAS; NISHIYAMA; RIBEIRO; FREITAS, 2016). Os genes CHD8 e KDM5C parecem conectar o componente genético, ao componente epigenético. A ligação da genética e epigenética pode ocorreratravés do gene CHD8, esse gene é responsável por codificar uma helicase, enzima com função remodeladora da cromatina e regulação da transcrição. O gene CHD8 apresenta grande associação com o TEA e foi identificado por apresentar 59 mutações diferentes, sendo 29 delas de perda de função (FISCHBACH, LORD, 2010 apud FREITAS; NISHIYAMA; RIBEIRO; FREITAS, 2016). O gene KDM5C codifica uma Demetilase Lisina-específica 5C e tem 28 mutações, associadas com retardo mental, ligado ao X, além de fenótipo associado a TEA (FREITAS; NISHIYAMA; RIBEIRO; FREITAS, 2016). Outros genes em que foi verificado uma forte associação com o TEA são o ADNP que é um gene localizado no cromossomo 20, o qual codifica uma proteína envolvida na remodelação da cromatina, na autofagia e na dinâmica dos microtúbulos nos sítios de sinapse e nas células da glia. Foram descritas mutações no gene ADNP em 10 pacientes com TEA. O gene TBR1 é um gene localizado no cromossomo 2, o qual codifica uma proteína que funciona como fator de transcrição, as alterações desse gene estão associados a enfermidades como Alzheimer e Parkinson (REYNOSO; RANGEL; MELGAR, 2015). Por: Jéssica Albino, Enfermeira e Acadêmica do curso de Medicina da UNILA. REFERÊNCIAS: FREITAS, P. M. de.; NISHIYAMA, P. B.; RIBEIRO, D. O.; FREITAS, L. M de. Deficiencia intelectual e o transtorno do espectro autista: fatores genéticos e neurocognitivos. Pedagogia em ação. v.8, n.2, 2016. COUTINHO, J. V. S. C.; BOSSO, R. M. do V. Autismo e genética: uma revisão de literatura. Revista Científica do ITPAC. v.8, n.1, 2015. REYNOSO, C.; RANGÉL, M. J.; MELGAR, V. El transtorno del espectro autista: aspectos etiológicos, diagnósticos y terapêuticos. Rev Med Inst Mex Seguro Soc. v. 55., 2017. |
