Dúvidas sobre
doenças genéticas?
Pergunte que eu respondo!

 A síndrome de Hutchinson-Gilford (HGPS) é uma doença genética rara (com uma prevalência de 1 em 20 milhões) caracterizada por manifestações semelhantes ao envelhecimento precoce causada por mutações heterozigóticas no gene LMNA (que codifica lâmina do tipo A), com mais de 90% dos pacientes carregando uma mutação pontual c.1824C> T (pG608G). Esta mutação ativa o uso de um sítio doador de splice 5 'alternativo no exon 11 que resulta na deleção de 150 nucleotídeos do mRNA e a síntese de uma proteína truncada chamada progerina. Esta proteína aberrante se acumula no envelope do núcleo devido à farnesilação irreversível e causa alterações graves nas funções celulares principalmente como um fator que leva a distúrbios na organização da heterocromatina, mitose, replicação e reparo do DNA e transcrição gênica.
0 Comments
 O rastreamento de doenças genéticas faz parte do acompanhamento pré-natal de gestantes e a avaliação dependerá de diversos fatores, como a probabilidade do desenvolvimento de anormalidades genéticas e possíveis riscos no caso da realização de exames mais invasivos.
Dessa maneira, existem uma série de exames que podem ser solicitados pelo médico obstetra, ou geneticista, quando for o caso, para o diagnóstico pré-natal de doenças genéticas. São eles: Ultrassonografia pré-natal: Exame não invasivo e sem risco conhecido para a mãe ou bebê. No segundo ou terceiro trimestre pode identificar malformações de estruturas cranianas, rins, coração, coluna, ossos e etc. Quando a ultrassonografia é direcionada, pode-se identificar precocemente doenças como microcefalia, hérnia diafragmática e malformações renais como a Síndrome de Potter, por exemplo. Amniocentese: Comumente, indicada após a décima quarta semana, visto que antes desse período os riscos de aborto espontâneo são aumentados. Assim, esse exame apenas é indicado quando se há suspeitas de doenças genéticas a partir de exames anteriores ou histórico familiar. Dessa maneira, é coletado líquido amniótico e células fetais a partir de uma agulha inserida via transabdominal, para testes genéticos e medição de marcadores, por exemplo. Sendo possível acusar diversas doenças, tais como: anemia falciforme, distrofia muscular e fibrose cística, por exemplo. Amostragem do vilo corial: Aspira-se as vilosidades coriônicas por meio de uma seringa e após a cultura celular as células são analisadas. Esse exame fornece as mesmas informações da amniocentese, porém a biópsia do vilo corial é feita entre a décima semana e o fim do primeiro trimestre de gestação. Cariótipo fetal: A partir da coleta da amostra de sangue fetal (por amniocentese ou por biópsia do vilo corial) pode ser feito o cariótipo para a análise de alterações a nível de cromossomo. Pode-se investigar trissomias, por exemplo, como a Síndrome de Down, Síndrome de Patau, Síndrome de Edwards. Além desses exames, também são solicitados, tradicionalmente, como triagem do primeiro trimestre a dosagem de: Beta-hCG sérica materna, uma vez que altos níveis podem ser associados com a Síndrome de Down; medição da proteína do plasma A (PAPP-A), visto que baixos níveis dessa proteína estão relacionados com o aumento da translucência nucal (por ultrassonografia), indicando risco para Síndrome de Down e cardiopatias congênitas, por exemplo. Dessa maneira, esses são alguns exemplos de exames de rastreio de doenças genéticas realizados no período pré-natal para a identificação de doenças genéticas, ainda no período intrauterino, de acordo com o histórico familiar, avaliação de risco e peculiaridades de cada paciente. Por: Camila Begui, Bióloga e Acadêmica do curso de Medicina da UNILA. REFERÊNCIAS: XAVIER, J. Pré-natal é essencial para o diagnóstico precoce de doenças raras. FIOCRUZ, Rio de Janeiro, 2017. Disponível em: https://portal.fiocruz.br/noticia/pre-natal-e-essencial-para-o-diagnostico-precoce-de-doencas-raras. Acesso em: 3 de nov. de 2021. DUNGAN, J. Exames diagnósticos pré-natais. Manual MSD, Canadá, 2019. Disponível em: https://www.msdmanuals.com/pt/casa/problemas-de-sa%C3%BAde-feminina/detec%C3%A7%C3%A3o-de-dist%C3%BArbios-gen%C3%A9ticos/exames-diagn%C3%B3sticos-pr%C3%A9-natais?query=Avalia%C3%A7%C3%A3o%20gen%C3%A9tica . Acesso em 4 de nov. de 2021.  A síndrome de Klinefelter é uma condição cromossômica em meninos e homens que pode afetar o desenvolvimento físico e intelectual. Mais comumente, os indivíduos afetados são mais altos do que a média são incapazes de ter filhos biológicos (inférteis); porém os sinais e os sintomas da síndrome de Klinefelter variam entre meninos e homens com essa condição. Em alguns casos, as características da condição são tão leves que a condição não é diagnosticada até a puberdade ou idade adulta, e os pesquisadores acreditam que até 75 por cento dos afetados homens e meninos nunca são diagnosticados.
Como acontece essa síndrome? Dentro das nossas células (no núcleo), o nosso material genético está organizado na forma de cromossomos, onde temos 22 pares de cromossomos autossômicos e 2 denominados sexuais. A síndrome de klinefelter acontece quando há dois cromossomos X no indivíduo fenotipicamente masculino. O cariótipo mais comumente associado a síndrome de Klinefelter é 47 XXY Porém, pode haver alguns indivíduos mosaicos em que algumas células possuem um número adequado de cromossomas mas enquanto outros possuem um X supranumerário e em 90% dos casos o cariótipo da síndrome de Klinefelter é o 47 XXY. Geralmente a Síndrome de Klinefelter ocorre aleatoriamente devido a um erro na não-disjunção meiótica, esse erro pode acontecer tanto na gametogênese materna quanto na paterna. Os sintomas da síndrome da síndrome de Klinefelter geralmente do fenótipo clínico típico é de um homem de alta estatura, membros compridos, pequenos testículos, ginecomastia (aumento das mamas) e diminuição do número de espermatozoides. Também se associa a gravidade do fenótipo com o número de cromossomo X a mais. Em alguns casos a síndrome de Klinefelter só vai ser diagnosticada quando o casal foi procurar um médico para pesquisar infertilidade, sendo a síndrome diagnosticada em 3% dos homens inférteis. Alguns estudos têm demonstrado que o cromossomo X extra e o material que ele codifica é responsável pela hialinização e fibrose dos testículos levando a sintomas de falência que podem aparecer como puberdade atrasada e também como ginecomastia. O diagnóstico da síndrome de klinefelter é confirmado a partir da suspeita clínica pelo cariótipo, ao ser encontrado o cromossomo X a mais em uma linhagem celular ou em todas as células. A síndrome de Klinefelter infelizmente não tem cura, o tratamento é multidisciplinar para as várias complicações que essa síndrome pode ter. Suplementação com testosterona, com supervisão de um endocrinologista pediátrico pode ser usada em alguns casos, principalmente para diminuir as características físicas dessa síndrome, como por exemplo para aumentar o tamanho do pênis e virilização. A longo prazo indivíduos com síndrome de Klinefelter podem vir a desenvolver diabetes tipo 2, dislipidemia e esteatose hepática, além de doenças tromboembólicas, demonstrando a importância do seguimento correto e do diagnóstico. Por: Nicolas Guzman, Acadêmico do curso de Medicina da UNILA. FONTE: https://pubmed.ncbi.nlm.nih.gov/?term=%28Klinefelter+Syndrome%5BMA JR%5D%29+AND+%28Klinefelter+syndrome%5BTI%5D%29+AND+english%5Bla% 5D+AND+human%5Bmh%5D+AND+%22last+1800+days%22%5Bdp%5D  O desenvolvimento do câncer de mama está relacionado a inúmeros fatores, que deixam o indivíduos mais suscetível a ficar doente, são eles: aumento da idade, fatores endócrinos/história reprodutiva, fatores comportamentais/ambientais e fatores genéticos/hereditários (ADAMI et al., apud INCA, 2021).
No caso do câncer de mama, o histórico familiar constitui uma condição epidemiológica de alto risco, pois indica a existência de uma predisposição hereditária ao câncer de mama que englobam as decorrentes circunstâncias: familiares acometidos em três gerações contínuas; dois ou mais parentes de primeiro grau observados com a doença no ciclo. A ginecomastia, edema do tecido mamário masculino, proveniente de um desequilíbrio hormonal, é um fator de risco para o câncer de mama masculino (VIEIRA et al.; ZEITUNE et al. apud COELHO et al., 2018). Para que uma célula deixe de ser saudável e se transforme em uma célula neoplásica ocorrem diversas mutações alteram a grade da matriz, ou seja, quando há uma desregulação da função de genes que agem indiretamente ou diretamente na proliferação ou na sobrevida das células, como os genes supressores de tumor e proto-oncogenes. Os oncogenes mais comuns são alelos mutantes de uma classe de genes celulares normais. São conhecidos como proto-oncogenes e seu funcionamento ou expressão, quando alterados, resultam em estimulação anormal da divisão celular e proliferação (AMENDOLA; VIEIRA apud COELHO et al., 2018). A presença de apenas um alelo com mutação já pode modificar o fenótipo de uma célula normal para maligno. Em contrapartida, os genes supressores tumorais, como por exemplo os genes BRCA1 e BRCA2, quando estão alterados, ocorre um mecanismo de perda de função de ambos os alelos, ou seja, um alelo mutante é herdado e o segundo alelo é inativado por um evento somático. Ademais, genes supressores tumorais, quando mutados, suprimem os chamados genes protetores (gatekeepers) que são responsáveis por regular diretamente o crescimento celular (DUFLOTH et al.; KERR; ASHWORTH; MACLEOD; NUSSBAUM; MCINNES; WILLARD apud COELHO et al., 2018). A predisposição genética a tumores é mediada pela herança da inativação de genes supressores de tumores, particularmente em famílias de alto risco. Os mais importantes genes supressores de tumor associados com o câncer de mama são os genes BRCA1 e BRCA2. Mulheres portadoras de mutações no BRCA1 possuem até 80% de chance de desenvolver câncer de mama, e até 60% de desenvolver câncer de ovário em sua vida (INCA; DUFLOTH apud COELHO et al., 2018) O gene BRCA1 está localizado no braço longo do cromossomo 17, constituído por 22 éxons codificantes, e codifica para uma proteína de 1.863 aminoácidos. Enquanto o gene BRCA2 se encontra no braço longo do cromossomo 13, sendo composto de 27 éxons codificantes, dos quais 26 codificam uma proteína com 3.418 aminoácidos. Esses dois genes são incumbidos de codificar proteínas nucleares expressas que são conservadas com integridade genômica por regular o reparo de DNA. Sua função é coibir o desenvolvimento de tumores por meio do reparo de DNA que estão alterados. Assim, as proteínas que são codificadas pelos genes interagem com outras proteínas para reparar as quebras de DNA. As proteínas BRCA1 e BRCA2 também são responsáveis por organizar a atividade de outros genes e também exercem um papel importante para o desenvolvimento embrionário (ECONOMOPOULOU; DIMITRIADS; PSYRRI; CARDOSO; FAGANELLO; LAJUS; AMENDOLA; VIEIRA apud COELHO et al., 2018). Os fatores genéticos/hereditários foram relacionados também à presença de mutações em outros genes como: PALB2, CHEK2, BARD1, ATM, RAD51C, RAD51D e TP53. Mulheres que possuem vários casos de câncer de mama e/ou pelo menos um caso de câncer de ovário em parentes consanguíneos, sobretudo em idade jovem, ou câncer de mama em homem também em parente consanguíneo, podem ter predisposição hereditária e são consideradas de risco elevado para a doença. O câncer de mama de caráter hereditário corresponde, por sua vez, a apenas 5% a 10% do total de casos (BREAST CANCER ASSOCIATION CONSORTIUM; GARBER et al.; ADAMI et al., apud INCA, 2021). Por: Jéssica Albino, Enfermeira e Acadêmica do Curso de Medicina da UNILA. REFERÊNCIAS BRASIL. Instituto Nacional do Câncer - Inca. Ministério da Saúde. Controle do câncer de mama: fatores de risco. 2021. Disponível em: https://www.inca.gov.br/controle-do-cancer-de-mama/fatores-de-risco. Acesso em: 14 out. 2021. COELHO, Aline Silva; SANTOS, Marielle Anália da Silva; CAETANO, Rosecleide Inácio; PIOVESAN, Camila Fátima; FIUZA, Larissa Aparecida; MACHADO, Ricardo Luiz Dantas; FURINI, Adriana Antônia da Cruz. Hereditary predisposition to breast cancer and its relation to the BRCA1 and BRCA2 genes: literature review. Revista Brasileira de Análises Clínicas, [S.L.], v. 50, n. 1, p. 1-7, abr. 2018. Revista Brasileira de Analises Clinicas. http://dx.doi.org/10.21877/2448-3877.201800615. 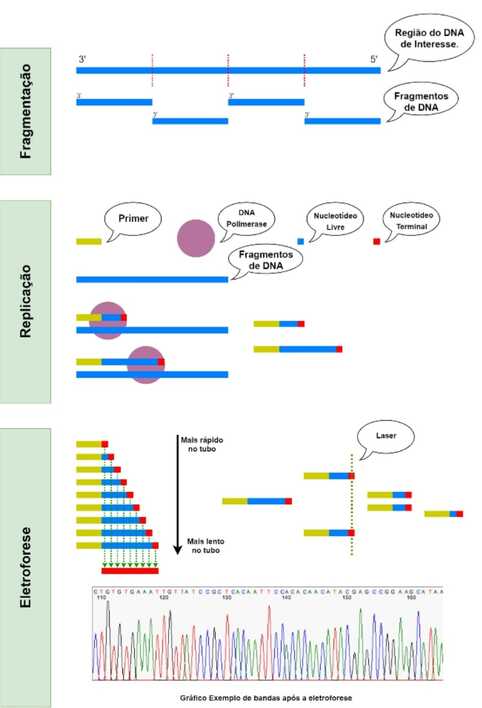 Essa postagem tem por intuito ser a primeira de uma série de 3 postagens falando sobre técnicas de sequenciamento genético. Neste primeiro artigo é abordado os eventos que ocorrem durante o sequenciamento usando o Método de Sanger.
Com os avanços dos conhecimentos sobre o material genético provocados pelas descobertas de Watson e Crick, ao final do século XX surgiu a necessidade de compreender conceitos além do “formato” da molécula carregadora de informações. Foi então em 1977 que Frederick Sanger e seus associados desenvolveram um método capaz de encontrar a ordem de pares de bases para um certo fragmento de uma molécula de DNA. Essa nova tecnologia de sequenciamento genético (o “Sequenciamento Sanger”) era única no mercado e, consequentemente, se tornou a com o mais amplo uso na atualidade. Para aplicação do Método de Sanger é preciso, primeiro, realizar uma preparação da amostra, que é a região do DNA de interesse a ser sequenciado. Esse preparo consiste em três partes principais: a fragmentação, a multiplicação e o isolamento. Na parte de fragmentação, a amostra é fragmentada em partes menores, as quais serão protagonistas durante o sequenciamento. Esses pequenos fragmentos, então, são separados e multiplicados, no intuito de aumentar a quantidade de material amostral. Um dos processos de multiplicação mais usados é a inserção destes fragmentos em plasmídeos de bactérias, as quais vão se proliferar através de clonagem. Por fim, após apresentar-se a quantidade de material desejada, é necessário isolar o material genético dos outros componentes usados para a fragmentação e proliferação. Dessa forma, cada secção da molécula de DNA apresenta, agora, em grande quantidade; obtendo-se, assim, amostras prontas para a análise. Após o preparo, uma das amostras (um dos grupos de fragmentos da molécula de DNA maior) é inserida em um ambiente rico em nucleotídeos e componentes para replicação (primers e DNA Polimerases). Por haver vários componentes para a replicação e pequenos fragmentos idênticos de DNA, inicia-se o processo de replicação. No entanto, ocorre a presença de dois tipos de nucleotídeos: os livres, que são nucleotídeos comuns, semelhantes aos usados em uma replicação em condições normais; e os marcados com material fluorescente, que, além da presença desse material, também apresenta uma alteração na ribose (substituição dos grupos hidroxila por hidrogênios) que permite com que ele não possa realizar ligação fosfodiéster, o que permite que sejam chamados de nucleotídeos terminais. Semelhante ao PCR, ciclos de temperatura são usados para a realização do processo de sequenciamento. Inicia-se com um aumento de temperatura para separar as duas moléculas de DNA da fita dupla durante o preparo da amostra. Já em uma temperatura menor, ocorre a aproximação do primer e da DNA Polimerase. Nesse momento, são depositados nucleotídeos na sequência adequada para o fragmento sendo analisado, no entanto de forma aleatória entre os nucleotídeos livres e terminais. Ao ser depositado um nucleotídeo terminal, a DNA Polimerase para de realizar a replicação, já que não é possível continuar o processo sem continuar fazendo ligações fosfodiéster. Dessa forma, a partir do fragmento analisado, fragmentos menores e de diferentes tamanhos são formados, com seus nucleotídeos finais estando marcados pelo material fluorescente. Então, é realizado uma eletroforese com o conteúdo resultante das reações da replicação. A eletroforese consiste na passagem dessas moléculas por um tubo com gel a partir de movimentos eletromagnéticos, devido a diferença de potencial entre as extremidades do tubo. Também, em um ponto mais distante no percurso do tubo, é introduzido um Laser capaz de estimular a marca fluorescente dos nucleotídeos terminais. Nesse ponto, devido ao atrito entre as moléculas de DNA e o gel, as moléculas menores percorrem a extensão do tubo mais rapidamente, ficando a frente, enquanto as mais longas ficam por último – ativando, assim, o laser em tempos diferentes. Portanto, uma câmera capta a luz resultante da marca dos nucleotídeos terminais, a qual se diferencia entre ATCG através de cores – como A sendo verde, T azul, C vermelho e G amarelo, por exemplo. É por conta da diferença de tamanho que é possível determinar a ordem dos nucleotídeos terminais que durante a eletroforese. Isso porque, uma molécula de DNA com um primer e um nucleotídeo terminal, ocuparia a vanguarda do percurso do tubo; sendo, assim, o nucleotídeo correspondente ao primeiro da sequência do DNA analisado. O segundo, seria um nucleotídeo mais longo (apresentando um primer, um nucleotídeo polimerizado do tipo livre e outro nucleotídeo polimerizado do tipo terminal), o terceiro 2 mais longo e assim sucessivamente. Isso permite, portanto, determinar a ordem de nucleotídeos em uma molécula de DNA. Por: Freddy Romanno Aires Nader, Acadêmico do curso de Medicina da UNILA  A variabilidade genética refere-se as diferenças entre indivíduos ou diferenças entre populações. As mutações são a causa de base da variabilidade genética, mas mecanismos como a reprodução sexual e à deriva genética também contribuem para isso. A variabilidade no genoma humano pode se apresentar de diferentes maneiras uma delas são os polimorfismos e em outras ocasiões, as variações ocorrem em uma escala maior, por exemplo, quando um segmento de DNA de centenas ou milhares de pares de bases é diferente entre as pessoas.
O DNA de todas as espécies conhecidas de organismos tem a mesma estrutura química; entretanto, cada organismo é completamente diferente do outro; a diferença se deve à ordem das bases nitrogenadas na molécula de DNA. Organismos da mesma espécie compartilham sequências em sua molécula de DNA, mas, mesmo dentro da mesma espécie existem variações entre os indivíduos. Organismos da mesma espécie compartilham regiões de sua sequência em até pouco mais de 99%, o que lhes confere características muito semelhantes. Além disso, parentes próximos terão sequências com maior similaridade entre si, mas nunca serão as mesmas, o que define a variabilidade genética intra e interespécies. No caso do DNA humano, as sequências que contêm os genes não são muito variáveis dentro da espécie; No entanto, o restante da sequência é muito sujeito à variabilidade e, como existem milhões de pares de bases por molécula de DNA e uma alta porcentagem deles codificam alguma proteína, cada pessoa tem uma sequência única de DNA o que permite que isso aconteça identificado apenas pela ordem de seus pares de bases. O que são os polimorfismos? Polimorfismo significa literalmente "muitas formas", se a variação é encontrada em uma frequência superior a 1% da população, se denomina polimorfismo. Os polimorfismos são variantes do genoma que aparecem por meio de mutações em alguns indivíduos e são transmitidos aos descendentes pelo que adquirem certa frequência na população após múltiplas gerações. Estima-se que exista uma variante em cada 1.000 pares de bases dos 3.000 milhões que constituem o genoma humano. Os polimorfismos são a base da evolução e aqueles que se consolidam podem ser silenciosos ou trazer vantagens para os indivíduos, embora também possam contribuir para causar doenças. Muitas doenças determinadas geneticamente pelas chamadas mutações ou variantes de "alta penetrância" são conhecidas, uma vez que os portadores da variante tendem a manifestar a doença com alta probabilidade. Assim, o polimorfismo genético, cromossômico ou de sequência de DNA são responsáveis pela grande variabilidade existente entre indivíduos de uma mesma espécie. Eles podem atuar como marcadores genéticos, já que são transmitidos e associados a outros genes localizados na região cromossômica próxima a eles. Desta forma, se um gene próximo a um marcador causa uma doença, todos os indivíduos afetados na família recebem tanto o marcador como o gene causador da doença. Essas variantes tendem a ser de baixa frequência na população em geral, por exemplo, mutações herdadas no gene supressor de tumor APC determinam o aparecimento de Polipose Adenomatosa Familiar que muitas vezes evolui em carcinoma no cólon, mas essa entidade não explica mais do que 1% de todos os tumores de cólon. Além disso, diferentes fenótipos são decorrentes de alguns polimorfismos, como, por exemplo, o sistema ABO. Os polimorfismos podem ter significados funcionais diferentes, dependendo se eles afetam uma região codificadora do genoma, uma região reguladora ou uma região não codificadora. Os polimorfismos nas regiões codificantes são chamados de "polimorfismos genéticos". Esta classe pode ou não ter efeito sobre o fenótipo. Polimorfismos gênicos, sem efeito fenotípico, são os mais comuns e são responsáveis pela diversidade genética normal entre os indivíduos (por exemplo, polimorfismos existentes em proteínas plasmáticas, como imunoglobulinas). Mas quando um polimorfismo gênico (ou seja, um gene afetado na região do DNA codificador) resulta em uma alteração fenotípica, na maioria das vezes é prejudicial, pois pode modificar as características bioquímicas, fisiológicas e até morfológicas da célula, podendo originar processos patológicos. Apenas em casos excepcionais, essa variação ou mutação pode ser benéfica, dando origem a uma vantagem adaptativa ao indivíduo, sendo este o motor da evolução da espécie. Polimorfismos com alteração do fenótipo, mas que não influenciam a susceptibilidade a doenças, determinam as características diferenciais entre indivíduos de uma mesma espécie, como altura, cabelo e cor dos olhos, etc. Polimorfismos para fins diagnósticos A análise dos polimorfismos pode ser utilizada para detectar a predisposição a uma determinada doença, e até mesmo detectá-la antes de seu desenvolvimento. Isso é de grande interesse no diagnóstico pré-natal de doenças congênitas. A identificação precoce desse tipo de doença pode permitir o início de um tratamento preventivo o que é mais eficaz para atenuar as manifestações clínicas da doença em questão. Por outro lado, é possível estudar á herança e a relação das doenças genéticas com as famílias, e está sendo utilizada em doenças como fibrose cística e coreia de Huntington. A impressão digital genética pode ser utilizada em estudos para estabelecer relações de parentesco, em testes de paternidade, na identificação de recém-nascidos e em casos de criminologia, entre outros. O princípio básico deste método é conseguir examinar um número suficiente de polimorfismos, o que gera uma probabilidade menor de que um outro indivíduo tenha os mesmos alelos na sequência de DNA, logo comparam as amostras para encontrar por exemplo o responsável de um crime. Por: Catherine Molina, Acadêmica do curso de Medicina da UNILA. Referências: Herrera, Edwin. La genética de poblaciones y el origen de la diversidad humana. Revista médica de Honduras. Vol 81 (1), 2013. Disponível em: http://www.bvs.hn/RMH/pdf/2013/pdf/Vol81-1-2013-10.pdf Iniesta, Raquel; Guinó, Elisabet; Moreno, Víctor. Análisis estadístico de polimorfismos genéticos en estudios epidemiológico. Revista Gaceta Sanitaria. Vol.19 (4), Barcelona, España, 2005. Disponível em: https://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S0213-91112005000400011 Thompson, Margaret; Thompos, James. Genética Médica. Editorial Elsevier. 6° Edição, 2002. Torrades, Sandra. Diversidad del genoma humano: polimorfismos. Elsevier. Vol 21 (5), mayo 2002. Págs. 122-125. Disponível em: https://www.elsevier.es/es-revista-offarm-4-articulo-diversidad-del-genoma-humano-polimorfismos-13031745 As mitocôndrias são organelas celulares que possuem DNA próprio. Como via de regra, para os seres humanos, considera-se que a herança mitocondrial seja materna, ou seja, o DNA presente nas mitocôndrias (mtDNA) é herdado da mãe, visto que as mitocôndrias do espermatozoide são eliminadas no processo de fertilização e somente aquelas presentes no oócito são conservadas no zigoto (YAN, C. et al. 2019). No entanto, contrariando o dogma central da herança mitocondrial, os estudos de Luo et al. (2018), apontam para casos excepcionais, em que esse padrão de herança pode não ocorrer, necessariamente: a heteroplasmia. A heteroplasmia consiste na herança biparental do mtDNA, ou seja, o indivíduo apresenta tanto o genoma mitocondrial materno quanto o paterno, fazendo com que o padrão de herança seja afetado, a depender da análise do tecido em questão e da porcentagem expressa de mtDNA em cada indivíduo, interferindo categoricamente no aconselhamento genético, sendo que estudos pontuais, individuais e altamente refinados serão necessários para a análise de cada caso. Dessa maneira, a heteroplasmia foi identificada pelos pesquisadores ao estudar indivíduos suspeitos de sofrerem distúrbios mitocondriais. Assim, ao realizar o sequenciamento do material genético mitocondrial, puderam observar a presença do mtDNA de ambos os pais, expressa nos filhos e, inclusive, ao longo de toda a família, caracterizando essa herança como autossômica dominante. De acordo com os resultados, o heredograma a seguir, retirado do estudo em questão, apresenta o padrão de herança heteroplásmica de uma das famílias estudadas: 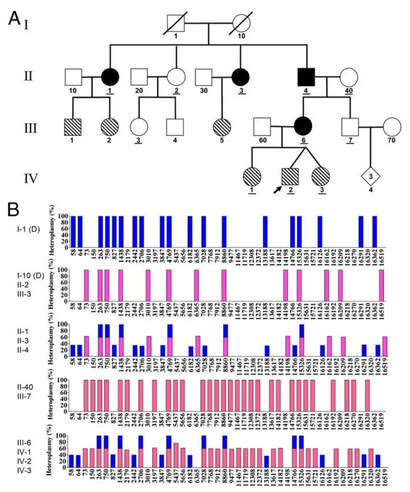 Luo et al. (2018) – modificado. No esquema A, os símbolos preenchidos em preto são os indivíduos que receberam a herança biparental mitocondrial. Já os preenchidos na diagonal indicam os indivíduos com alto nível de heteroplasmia, mas com herança materna normal. Os indivíduos sublinhados são os que foram sequenciados para o mtDNA e as entradas com “D” são genomas deduzidos. No esquema B, as barras azuis representam o genoma mitocondrial de origem paterna, já as barras em rosa e em vermelho, de origem materna.
Dessa forma, pode-se observar que o probando (IV-2) apresenta o mesmo mtDNA da mãe (III-6), a qual possui a heteroplasmia, visto que recebeu o mtDNA tanto de seu pai (II-4), quanto de sua mãe (II-40). Além disso, o heredograma também aponta para que esta seja uma herança autossômica dominante, na qual, uma vez que instalada, não tende a pular gerações. A partir do exposto, pode-se perceber que a herança mitocondrial se apresenta, na verdade, de uma forma muito mais complexa do que o clássico padrão apresentado até então. Cabe aos pesquisadores, portanto, compreender a gama de informações que vem se apresentando, para que no futuro, novas técnicas de reconhecimento, terapia e tratamento possam ser desenvolvidas à luz dos novos conhecimentos adquiridos, podendo vir a corroborar, inclusive, nos processos de aconselhamento genético que envolvam as doenças de origem mitocondrial. Por: Camila Begui, Bióloga e Acadêmica do curso de Medicina da UNILA. REFERÊNCIAS: LUO, S. et al. Biparental Inheritance of Mitochondrial DNA in Humans. Proceedings of the National Academy of Sciences, v. 115, n. 51, p. 13039-13044, dez. 2018. Disponível em: https://www.pnas.org/content/115/51/13039. Acesso em: 24 de agosto de 2021. YAN, C. et al. Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells, v. 8, n. 4, p. 379-394, abr. 2019. Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6523345/. Acesso em: 24 de agosto de 2021. Existe uma mutação genética descoberta no ano 1994 chamada: Fator V de Leiden. Essa mutação predispõe os portadores ao tromboembolismo hereditário, ao qual gera uma desordem multifatorial que envolve a interação de fatores de risco genéticos e/ou adquiridos que afetam as proteínas do sistema anticoagulante. Dentre esses fatores, as mutações nos genes do fator V e da protrombina são as duas causas que prevalecem para o desenvolvimento da trombose hereditária.
Para entendermos um pouco melhor, é importante explicar o que é o fator V: é uma proteína plasmática formada por 2.196 aminoácidos, precursor do fator Va (fator V ativado) essencial para a síntese da trombina, responsável pela ativação da coagulação. O processo de desativação baseia-se na ligação da trombina e da proteína C à trombomodulina (presente no endotélio) convertendo a proteína C à proteína C ativada (PCA). Esta ativação ocorre na presença da proteína S (PS). O complexo PCA/ PS causa a inativação através da clivagem enzimática dos cofatores Va e VIIIa que inibem a atividade do sistema coagulante. A resistência à proteína C ativada é um dos principais fatores de risco para o tromboembolismo venoso e é a mutação do fator V de Leiden que representa uma das causas principais de resistência à proteína C. Cerca de 90% dos casos de resistência a esta proteína se devem à mutação de ponto no gene do fator V, da coagulação. Esta mutação ocorre no exon 10 do gene do fator V ocasionando uma substituição da base G/A (Guanina/Adenina) no nucleotídio 1691, resultando na troca da Arg (Arginina) pela Gln (Glutamina) na posição 506 da proteína, um dos principais sítios de clivagem para ativação da proteína C. A presença da mutação aumenta o risco de doença trombótica de três a dez vezes para portadores heterozigotos e de oitenta vezes para portadores homozigotos. Estudos mostram que a prevalência desta mutação dependerá da etnia da população estudada. A taxa da mutação dentro da população européia é de 5%, nos Estados Unidos é de 6% e praticamente ausente entre os africanos e asiáticos. Em afro-americanos é encontrada em cerca de 1%. No Brasil, a mutação está presente em cerca de 2% da população. Em pacientes portadores de doenças trombóticas, o fator V de Leiden foi observado em 20% dos casos. As técnicas de biologia molecular tornam possível a detecção de mutações, que podem ser realizadas, atualmente, como rotina laboratorial. A aplicação de teste de rastreamento destas mutações em pessoas com antecedentes clínicos e/ou familiar são de grande importância, pois permitem uma abordagem clínica antitrombótica, de modo a diminuir os riscos destas doenças. Os pacientes com doenças trombóticas (trombose venosa profunda, embolia pulmonar, trombose arterial e trombose cerebral vascular), podem ser analisados juntamente com sua família para detecção da distribuição dos alelos mutados a fim de correlacionar a presença da mutação e a sintomatologia. Finalmente, a detecção do fator V de Leiden em pacientes portadores de eventos trombóticos é recomendada para o esclarecimento das causas e para efetuar o rastreamento em membros de sua família, ainda sem o aparecimento de eventos trombóticos, de forma a avaliar os riscos associados e assim determinar um acompanhamento médico preventivo. Por: Nicolas Guzman, Acadêmico do curso de Medicina da UNILA. REFERÊNCIAS: Carvalho, Eunice B. et al. Rastreamento familiar do fator V de Leiden: a importância da detecção de portadores heterozigotos. Revista Brasileira de Hematologia e Hemoterapia [online]. 2005, v. 27, n. 2, pp. 83-86. Disponível em: <https://doi.org/10.1590/S1516-84842005000200005>. Epub 11 Jan 2006. ISSN 1806-0870. https://doi.org/10.1590/S1516-84842005000200005. Godoy, José M. P.Fator V de Leiden. Revista Brasileira de Hematologia e Hemoterapia [online]. 2005, v. 27, n. 2, pp. 79. Disponível em: <https://doi.org/10.1590/S1516-84842005000200001>. Epub 11 Jan 2006. ISSN 1806-0870. https://doi.org/10.1590/S1516-84842005000200001  O Transtorno do Espectro Autista (TEA) é um transtorno que engloba o comportamento e cognição, geralmente com início antes dos 3 anos de idade, afetando os domínios fundamentais da linguagem e desenvolvimento social com comportamentos repetitivos e restritivos (COUTINHO; BOSSO, 2015). O TEA é classificado como multicausal, no entanto, existem duas vertentes principais do autismo: o sindrômico e não sindrômico.
Estudos familiares e em gêmeos evidenciam a etiologia genética do autismo, mostrando a existência de um risco aumentado de recorrência do autismo de 3 a 8% em famílias com uma criança autista e concordância para o diagnóstico de autismo em gêmeos monozigóticos de pelo menos 71% (SOLÍS- AÑES, 2007; DELGADO-LUENGO, 2007; HERNÁNDEZ, 2007; GARDIA, 2004; TUCHMAN, 2004; ROTTA, 2004 apud COUTINHO; BOSSO, 2015). Embora o autismo pareça ser altamente hereditário, sua etiologia genética tem se mostrado bastante complexa, provavelmente envolvendo muitos genes em diferentes cromossomos atuando com efeito moderado (SOLÍS-AÑEZ, 2007; DELGADO- LUENGO, 2007; HERNÁNDEZ, 2007; GESCHWIND, 2008 apud COUTINHO; BOSSO, 2015). Existem autores que chegaram a afirmar que anomalias de quase todos os cromossomos já foram associadas ao autismo. Não implicando em um modelo próprio de transmissão genética ou um gene principal facilmente identificável como causa de desordens (GESCHWIND,2008 apud COUTINHO; BOSSO, 2015). Estudos de genética humana indicam que os genes da família SHANK podem estar envolvidos no autismo idiopático ou não sindrômico. Mutações nesses genes causam uma disfunção sináptica, a qual leva ao comportamento autista. A primeira triagem do genoma para regiões cromossômicas envolvidas no autismo associou aproximadamente 354 marcadores genéticos, localizados em oito regiões dos seguintes cromossomos: 2, 4, 7, 10, 13, 16, 19 e 22 sendo as regiões 7q, 16p, 2q, 17q mais relevantes (COUTINHO; BOSSO, 2015). As regiões do genoma humano 6q27, 20p13, e 5p15 foram descritas como regiões associadas com o autismo (WEISS et al. 2009 apud FREITAS; NISHIYAMA; RIBEIRO; FREITAS, 2016). Os genes CHD8 e KDM5C parecem conectar o componente genético, ao componente epigenético. A ligação da genética e epigenética pode ocorreratravés do gene CHD8, esse gene é responsável por codificar uma helicase, enzima com função remodeladora da cromatina e regulação da transcrição. O gene CHD8 apresenta grande associação com o TEA e foi identificado por apresentar 59 mutações diferentes, sendo 29 delas de perda de função (FISCHBACH, LORD, 2010 apud FREITAS; NISHIYAMA; RIBEIRO; FREITAS, 2016). O gene KDM5C codifica uma Demetilase Lisina-específica 5C e tem 28 mutações, associadas com retardo mental, ligado ao X, além de fenótipo associado a TEA (FREITAS; NISHIYAMA; RIBEIRO; FREITAS, 2016). Outros genes em que foi verificado uma forte associação com o TEA são o ADNP que é um gene localizado no cromossomo 20, o qual codifica uma proteína envolvida na remodelação da cromatina, na autofagia e na dinâmica dos microtúbulos nos sítios de sinapse e nas células da glia. Foram descritas mutações no gene ADNP em 10 pacientes com TEA. O gene TBR1 é um gene localizado no cromossomo 2, o qual codifica uma proteína que funciona como fator de transcrição, as alterações desse gene estão associados a enfermidades como Alzheimer e Parkinson (REYNOSO; RANGEL; MELGAR, 2015). Por: Jéssica Albino, Enfermeira e Acadêmica do curso de Medicina da UNILA. REFERÊNCIAS: FREITAS, P. M. de.; NISHIYAMA, P. B.; RIBEIRO, D. O.; FREITAS, L. M de. Deficiencia intelectual e o transtorno do espectro autista: fatores genéticos e neurocognitivos. Pedagogia em ação. v.8, n.2, 2016. COUTINHO, J. V. S. C.; BOSSO, R. M. do V. Autismo e genética: uma revisão de literatura. Revista Científica do ITPAC. v.8, n.1, 2015. REYNOSO, C.; RANGÉL, M. J.; MELGAR, V. El transtorno del espectro autista: aspectos etiológicos, diagnósticos y terapêuticos. Rev Med Inst Mex Seguro Soc. v. 55., 2017. “O que nos faz ‘Homens’ ou ‘Mulheres’¿” Essa é uma pergunta que vem sendo cada vez mais realizada pelas pessoas na contemporaneidade. Por muito tempo acreditava-se que “aquilo que temos no meio das pernas” ditava o nosso sexo; depois passou-se a acreditar que o par de cromossomos sexuais determinavam com toda certeza cabível qual é meu sexo. Porém, relatos e estudos demonstram que a genética sexual é mais complexa do que apenas dois cromossomos no final do nosso cariótipo.
A advogada americana Kimberly Zieselman relata que aos 15 anos, por não poder menstruar, foi diagnosticada com câncer e que seu útero e ovário precisavam ser removidos. Kimberly passou pelo procedimento, e por isso precisou ficar em tratamento hormonal pelo resto da vida. Foi apenas aos 41 anos que ela e seus pais descobriram que o diagnóstico de câncer era falso. Na verdade, ela tinha uma chace de ter câncer, devido aos seus testículos internos. Esse e inumeros outros relatos, apesar de acompanhar uma grande carga emocional, retratam como pessoas que nasceram XY, mas apresentam atributos primários e características sexuais secundárias femininas, existem. E hoje movimentam estudos sobre como ocorre a definição sexual humana. Apesar de ainda não nos fornecerem toda a informação sobre o assunto, esses estudos já buscam concluir que o Sexo é algo muito mais complexo do que se imaginava. A ideia de sexo binário (a existência sumária de homens e mulheres apenas) começa a ser deteriorada à medida que os resultados desses estudos vão se apresentando. Isso começa a formar uma ideia de “sexo espectral”, em que existem intermediários entre o “macho” e a “fêmea” na espécie humana, tal qual ocorre com outros atributos (como coloração da pele, cor dos olhos, formatos do nariz e altura). Um estudo aponta como a diferenciação sexual acontece no cérebro a partir de mudança no formato do material genético provocada pela presença ou ausência de hormônios. Nesse estudo, de Matsuda, foi realizada a inibição do gene responsável por originar os receptores de Estrogênios (ERα e Erβ), no intuito de entender como esse gene influe sobre a diferenciação sexual. Os resultados não foram nada surpreendentes para as pesquisadoras, mas para nós pode vir como um choque. Esse gene ao ser inibido produzia ratos XY com comportamento masculino muito prejudicado. Em um outro estudo de 1990 por Sinclair, foi encontrado o principal responsável pela determinação do sexo masculino: o gene SRY no cromossomo Y. Nesse caso, mulheres que carregam parte do cromossomo Y com a presença desse gene podem apresentar características masculinas por si só. Já em um terceiro estudo, por Jordan, apontou que o cópias excessivas ou ausência do gene WNT4 tem grande influência sobre o fenótipo sexual do indivíduo. Esses estudos e vários outros mudaram o pensamento sobre a determinação sexual entre os animais. Em alguns isso já se faz muito bem conhecido (como nos ratos). Já nos humanos ainda existe uma grande jornada que precisa ser traçada para o entendimento do desenvolvimento desse aspecto. Porém, uma coisa é certa, a teoria vigente de que sexo é uma coisa estática e binária, começa a nos parecer tão defasada como a teoria do terraplanismo. Por: Freddy Romanno Aires Nader, Acadêmico do curso de Medicina da UNILA REFERÊNCIAS: Matsuda - Matsuda, K.I., Mori, H. & Kawata, M. Epigenetic mechanisms are involved in sexual differentiation of the brain. Rev Endocr Metab Disord 13, 163–171 (2012). https://doi.org/10.1007/s11154-012-9202-z Sinclair - Sinclair, A., Berta, P., Palmer, M. et al. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 346, 240–244 (1990). https://doi.org/10.1038/346240a0 Jordan - Jordan, B. K. et al. Am. J. Hum. Genet. 68, 1102– |
